 Welcome to the final article in our analysis and compliance in pharmaceuticals series. In this chapter we discuss systems for installation and analytical instrument qualification (AIQ) that ensure both compliance with pharma regulations and successful product development and manufacture.
Welcome to the final article in our analysis and compliance in pharmaceuticals series. In this chapter we discuss systems for installation and analytical instrument qualification (AIQ) that ensure both compliance with pharma regulations and successful product development and manufacture.
In the regulated environment of the pharmaceutical industry, commissioning and use of analytical instruments in accordance with the latest standards of the U.S. Food and Drug Administration (FDA) and GLP/GMP guidelines is mandatory in order to ensure accurate material analysis, consistent product manufacturing, and ongoing, verified monitoring of materials and procedures.
Metrohm has developed a modular system for the installation and qualification of instruments in strict accordance with the current regulations and provides Instrument Qualification (IQ), Operational Qualification (OQ) and Performance Verification (PV) documentation that satisfy USP 1058, GAMP, and FDA Title 21 CFR part 11 compliance. This chapter will provide a foundation for understanding the requirements of analytical instrument qualification (AIQ) to ensure you demonstrate traceable, guaranteed, and documented system performance.
Implemented in 2008, the United States Pharmacopeia (USP) is the only pharmacopeia with a chapter specific to AIQ. The new USP general chapter <1058> was released in August 2017 and defines AIQ as the collection of documented evidence that an instrument performs suitably for its intended purpose (1). This chapter will help you understand how USP <1058> defines and manages AIQ risk, why change control processes are important, and how integrated software facilitates compliance with 21 CFR Part 11.

Qualification Lifecycle
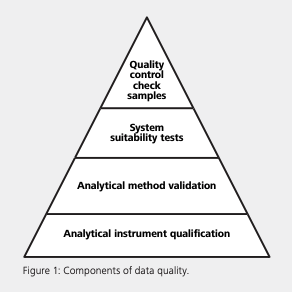
Data integrity is the fingerprint of a company’s processes and products. USP <1058> outlines four components within the quality triangle which are critical to data quality: AIQ, method validation, system suitability, and quality control checks (see Figure 1). While the AIQ contributes to confidence in the validity of data generated; method validation provides the proof that an analytical procedure is suitable for intended use. The system suitability test verifies that the system will perform in accordance with set criteria, and the quality control check provides ongoing assurance of suitable performance and accuracy. The base layer, AIQ, is expanded into the 4Q lifecycle phases for the defined specifications, installation assessment, and monitoring of ongoing instrument performance (Table 1) (1). While there are no regulatory barriers about who performs laboratory instrument qualification, the person performing the work must have the proper education, training, and experience.

Risk Assessment 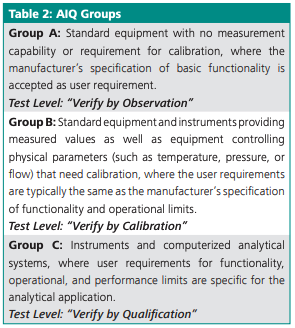
USP <1058> manages AIQ risk by classifying instruments based on complexity into three groups: A, B, or C (Table 2). Since the same instrument can exist in more than one group, depending on its use, risk assessment eliminates duplication of effort and avoids costly practices such as repeating qualification steps and generating redundant documentation (1). When an instrument’s intended use falls into Group C, all elements of qualification, including software validation, must be considered to ensure proper functioning of the instrument. Table 2 shows how an instrument can be grouped and which test strategy is therefore required to meet qualifications.

Change Control
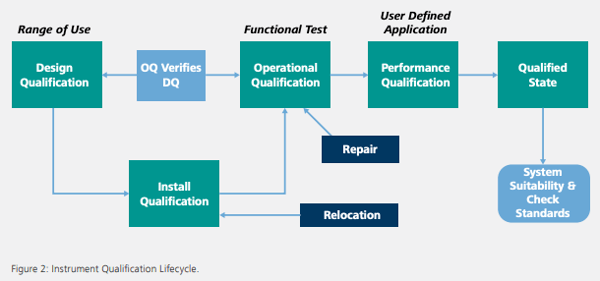
After impact has been established for the equipment or system, it is important to develop a change control process for lifecycle management. The change control process should include a documented SOP outlining requirements for instrument installation, failure management, routine maintenance and qualification frequency (Figure 2). Metrohm USA offers comprehensive IQ, OQ and PV packages that comply with the latest regulations and standards for easy implementation and liability protection. Metrohm USA recommends performing preventive maintenance and an operational qualification service annually to ensure reliable and accurate results. Key benefits of these packages include the following:
- Modular document structure
- Individual component tests
- Certified engineer and reference instruments
- Documented proof of whole system operation functionality
- Traceable monitoring of the system performance through regular requalification and testing
Software Compliance
The Title 21 Code of Federal Regulations Electronic Records; Electronic Signatures of the U.S. Food and Drug Administration (21 CFR Part 11) defines the requirements for electronic documentation and signatures. This rule, which has been in effect since August 1997, specifies how the system components, controls, and procedures have to be designed to ensure the reliability and authenticity of electronically stored records (2).
Achieving and maintaining full compliance with 21 CFR Part 11 necessitates standard operating procedures (SOPs) that support and complement the functionality of electronic systems. Products with integrated functions supporting 21 CFR Part 11 requirements ease the task of achieving and maintaining full compliance. Systems that include Metrohm’s tiamo™, MagIC Net, Mira Cal, Vision Air, and OMNIS software platforms have been developed from the ground up to satisfy these regulations (3).
Conclusion
Compliance with regulations in the pharmaceutical industry is essential to successful product development and manufacture. Employing systems that are designed to fulfill these requirements streamlines the process for ensuring complete, ongoing adherence to such regulations. Following guidelines for assessing instrument risk, developing change control processes, and using software designed around these regulations is key to ongoing success.

References
- United States Pharmacopeia General Chapter <1058> “Analytical Instrument Qualification”.
- http://www.fda.gov/ora/compliance_ref/Part11
- https://partners.metrohm.com/ GetDocument?action=get_dms_ document&docid=627230
